The CisCross web-server includes four modes: CisCross-Main, CisCross-Light, CisCross-TF-targets and CisCross-FindTFnet.
Mode 1 CisCross-Main implements the algorithm, described on Figure 1, to predict potential regulators of input set of genes.
Mode 2 CisCross-Light gives the list of DAP-Seq peaks detected in the 5’-regulatory region of the given input gene.
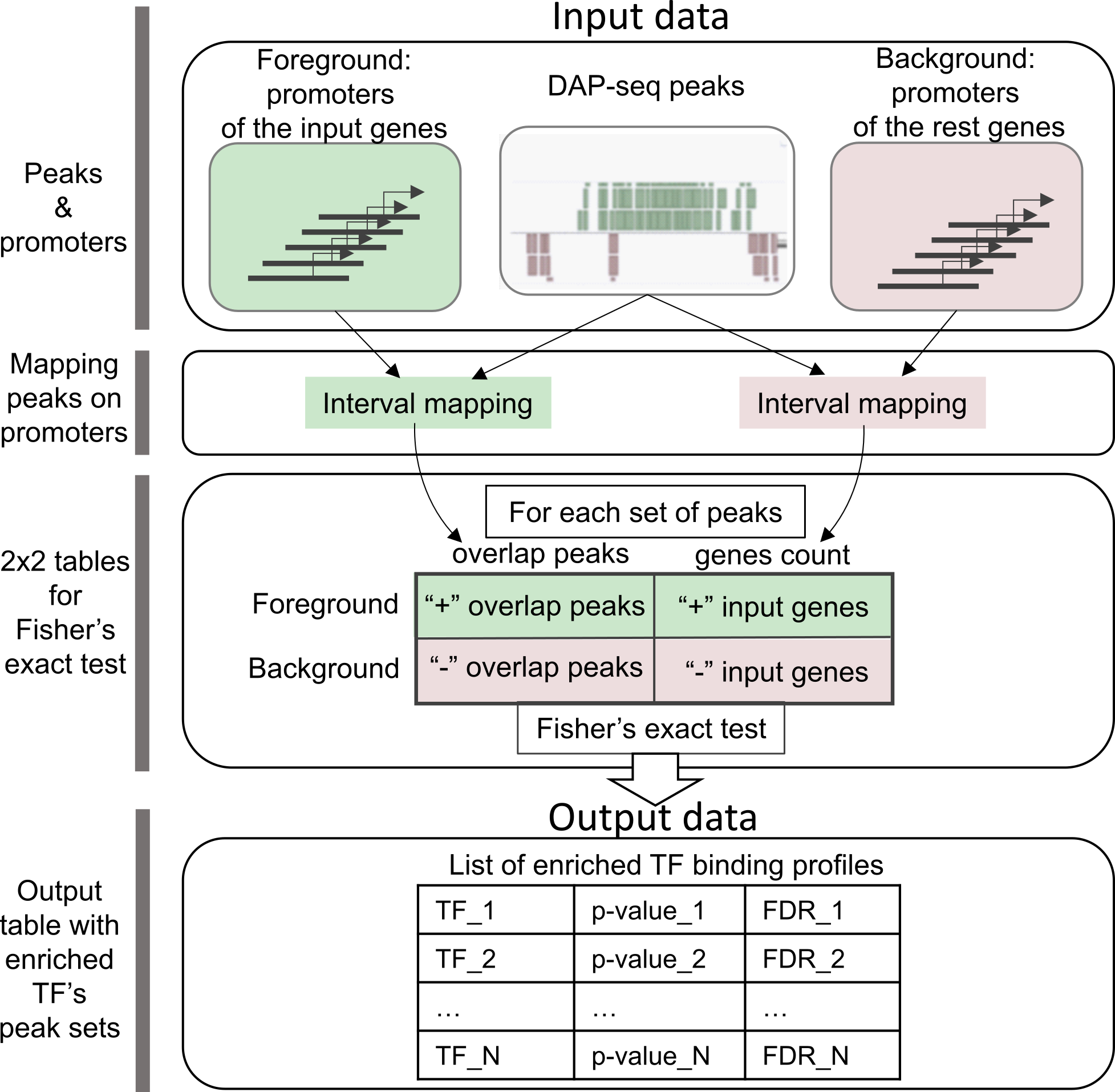
Figure 1. CisCross-Main algorithm scheme.
5’-regulatory regions of genes from an input list (for example, differentially expressed genes) are used as the foreground and those for the rest of Arabidopsis genes as the background. Green/pink colors mark foreground and background data and respective parallel processes of their analysis. Foreground and background data comprise the annotations of promoter regions of the input genes and the rest genes, respectively. For one DAP-seq set of peaks, the first step of the analysis maps the peaks to promoters of the input genes and the rest genes. For each TF peak set, CisCross counts the number of genes in the foreground/background, which 5’-regulatory regions overlap or do not overlap the TF binding peaks. The second step uses these data of genome mapping to compile a 2x2 contingency table for the input genes and the rest genes with the counts of genes whose promoters overlap or do not overlap the peaks. The significance p-value of the input gene promoter enrichment for the peaks is assessed using Fisher’s exact test. These calculations are performed for each set of peaks. Finally, CisCross runs through all the peak sets in the selected DAP-Seq collection and applies the correction for multiple testing to compute the False Discovery Rate (FDR) for each peak set. Output data comprise the list of enriched TF binding profiles with significance p-value in the ascending order of FDR (the significance p-value adjusted for multiple testing).
Download the re-processed peak sets MACS2, GEM
Download the results of de novo motif search
Mode 3 CisCross-TF-targets gives the list of target genes of TFs which DAP-Seq peaks detected in the 5’-regulatory region of the given input gene list.
Mode 4CisCross-FindTFnet implements a three-step procedure for the reconstruction of transcription factors regulatory networks (TFRNs), described on Figure 2, based on the analysis of upregulated and downregulated differentially expressed gene (uDEG and dDEG) lists.
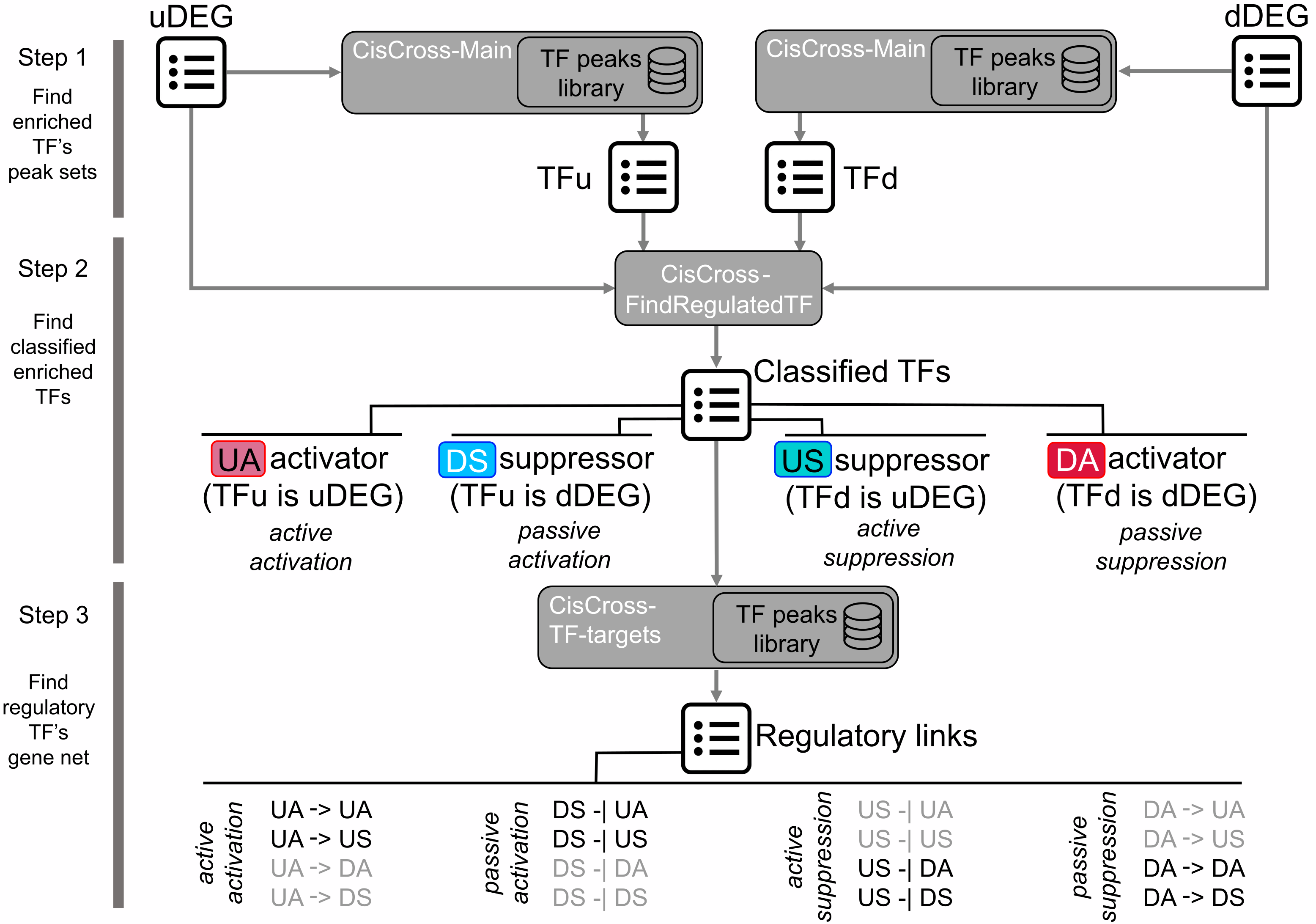
Figure 2. Reconstruction of induced TFRNs with FindTFnet.
A three-step procedure to infer “TF regulator–TF target” pairs.